computel的算法背景¶
computel这是一个用于计算整个基因组下一代测序数据中的平均端粒长度的R程序.它采用了基于短读取的比对方法,并集成了各种流行的测序数据分析工具。
computel以fastq文件作为输入,并从中估计相关样本的端粒长度。
下载,安装和运行computel以计算端粒长度¶
在此,我们将介绍如何下载computel,如何使用miniconda创建computel能够运行的环境,如何调试环境以让computel正确运行。
请务必注意,computel是基于R语言开发的,我们创建环境的时候需要创建R语言环境,否则无法正常运行computel。
# 创建R语言环境
conda create -n computel r
# 启动环境
conda activate computel
鉴于作者在官方GitHub仓库提供了相关代码的下载流程,我们在这里遵循作者的步骤来下载和安装Computel。
首先,我们从Computel的GitHub仓库中克隆Computel的代码。
# 克隆computel
git clone https://github.com/lilit-nersisyan/computel.git
我们需要将下载的Computel文件解压缩到本地目录。接着,安装所需的二进制文件和设置配置文件已经包含在软件包中。
为了使 computel.sh 文件可执行,我们使用 chmod +x computel.sh 命令。这是一个在Linux/Unix系统上用于更改文件权限的命令,其目的是授予 computel.sh 文件执行权限。
相关代码如下:
# 切换到computel目录
cd computel
# 更改computel.sh文件的权限
chmod +x computel.sh
# 运行computel.sh文件
sh computel.sh
当你成功安装computel之后,在linux命令行中会输出以下内容:
Program: computel Version: 1.3
usage: ./computel.sh [options] {-1
………… ………… …………
在运行 Computel 之前,我们需要安装 Computel 的相关依赖包,以确保 Computel 能够正常运行。
值得注意的是,我们必须使用 1.11 版本的 Samtools。如果您使用更高版本的 Samtools,可能会导致错误。这是因为 Computel 在设计时并没有考虑与更高版本的 Samtools 兼容。
# 安装computel依赖包bowtie2-build
conda install -c bioconda bowtie2
# 载入computel依赖包samtools
module load samtools/1.11
接下来,您可以使用以下简单的测试代码来验证是否成功安装了computel。
如果您在命令行中运行以下代码,能够正常输出下图所显示的结果,那么恭喜您,您已经成功安装了computel:
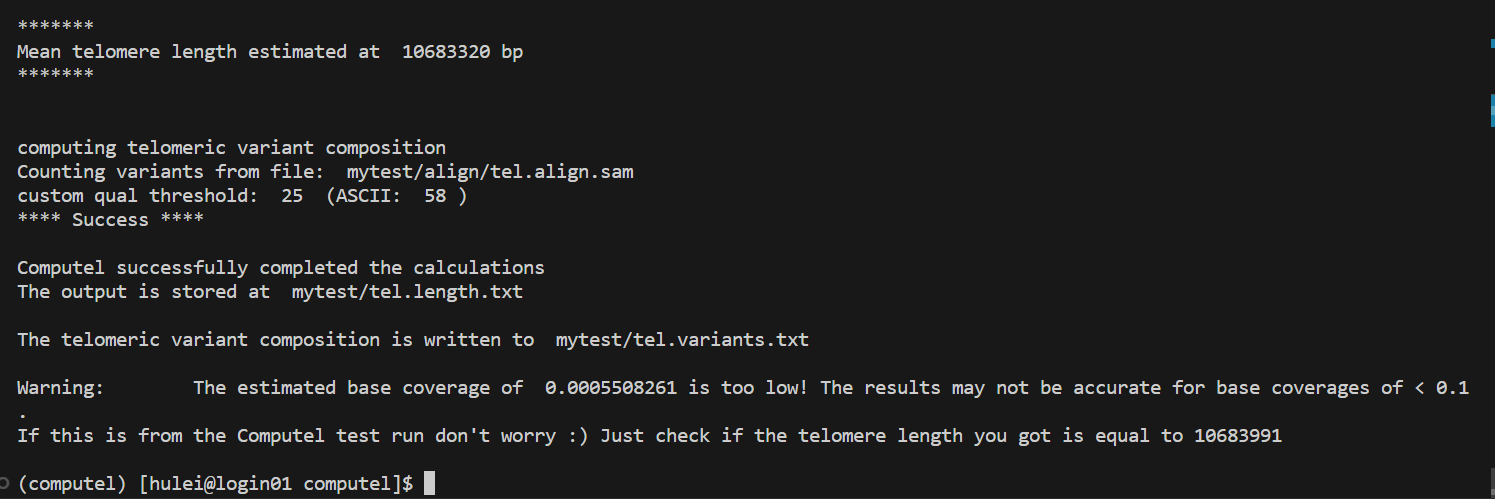
运行computel以估计样品的平均端粒长度¶
你可以使用命令行或西湖大学高性能计算平台的srun系统来运行computel。
下面提供了一个在srun系统中运行computel的模拟代码示例。在这个示例中,我们将计算特定路径下所有的fastq文件的平均端粒长度,并将结果输出到我们手动指定的目标文件夹中。
请注意,我们只需要指定使用computel计算端粒长度的样本名,输入fastq文件的目录,和保存输出结果的目标文件夹。
还请务必注意,在我的脚本中,我假定了fastq的样本名命名满足 ${file_name}_1.fastq.gz和 ${file_name}_2.fastq.gz格式,如果不满足这个格式,则无法正常运行代码。各位在使用我的代码时需要手动修改这里的路径和命名格式。
如果你想查找computel工具包的其他参数及其默认参数说明,你可以在computel文件夹下,在命令行中输入./computel.sh -h,即可得到相关参数说明。
#!/bin/bash
#SBATCH -p intel-sc3,amd-ep2
#SBATCH -q normal
#SBATCH -J preprocess
#SBATCH -c 6
#SBATCH -o /home/douyanmeiLab/hulei/WGS/telomere/Final_output/brain/computel.log
#SBATCH -t 120:00:00
#!/bin/bash
# 保存文件名的数组
file_names=("SRR6349071" "SRR6349073" "SRR6349107" "SRR6349108" "SRR6349116" "SRR6349117" "SRR6349211" "SRR6349213" "SRR6349138" "SRR6349140")
# 循环遍历文件名数组
for file_name in "${file_names[@]}"; do
# 构造输入文件路径
input_file1="/storage/douyanmeiLab/fanwenxuan/data/phs001485.v2.p1/fq/${file_name}_1.fastq.gz"
input_file2="/storage/douyanmeiLab/fanwenxuan/data/phs001485.v2.p1/fq/${file_name}_2.fastq.gz"
# 构造输出目录
output_dir="/home/douyanmeiLab/hulei/WGS/telomere/Final_output/brain/computel/${file_name}"
# 运行命令
sh /home/douyanmeiLab/hulei/WGS/telomere/computel/computel/computel.sh -1 "$input_file1" -2 "$input_file2" -o "$output_dir"
done
请注意,由于computel每次计算只能计算一个文件,且输出一个单独的文件夹,意味着如果你输入多个文件,computel的结果会保存在多个文件夹的tel.length.txt文件中,如下图所示,所以我们需要对srun系统中的运行代码进行修改,手动指定输出的文件路径。
computel结果说明和后处理¶
computel的输出结果是一个tel.length.txt文件。每个文件包含一列的index信息和一行的输出结果信息。
对于这种类型的文件,你可以在Linux中手动打开它,或者创建一个IPython Notebook文件,并使用Pandas库来打开txt文件。tel.length.txt文件的内容如下:
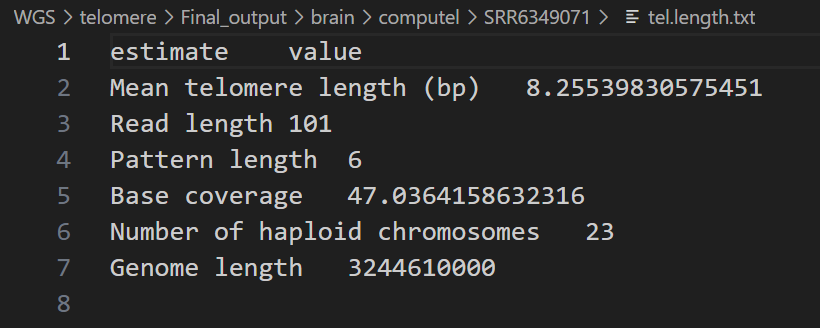
我们只需要关心输出文件中的Mean telomere length即可,这便是computel估计的端粒长度。
我们可以写一个python代码,手动读取目标文件夹下面所有computel的输出的tel.length.txt文件,然后将每个文件的输出结果合并在同一个tsv文件中,相关代码如下:
在这个代码中,我们只需要指定保存computel的输出结果的文件夹和保存合并后的tsv文件的路径即可。这个代码会手动合并所有结果。
import os
import pandas as pd
# 目标文件夹路径
target_folder = "/home/douyanmeiLab/hulei/WGS/telomere/Final_output/brain/computel"
output_file = "/home/douyanmeiLab/hulei/WGS/telomere/Final_output/brain/computel/merged_summary.tsv"
# 初始化一个空的DataFrame来存储合并的数据
merged_data = pd.DataFrame()
# 遍历目标文件夹及其子文件夹
for sample_folder in os.listdir(target_folder):
sample_path = os.path.join(target_folder, sample_folder)
if os.path.isdir(sample_path):
for root, dirs, files in os.walk(sample_path):
for file in files:
if file.endswith("tel.length.txt"):
file_path = os.path.join(root, file)
# 获取样本名(文件夹名)
sample_name = sample_folder
# 读取每个子文件夹中的_summary.tsv文件内容
data = pd.read_csv(file_path, delimiter='\t', index_col=[0])
data = data.T
# 添加ReadGroup列,内容为样本名
data['ReadGroup'] = sample_name
merged_data = pd.concat([merged_data, data], axis=0)
# 保存合并后的数据到一个新的tsv文件
merged_data.to_csv(output_file, sep='\t', index=False)
print(f"合并完成,合并后的数据保存在{output_file}中。")
代码的输出结果如下图所示:
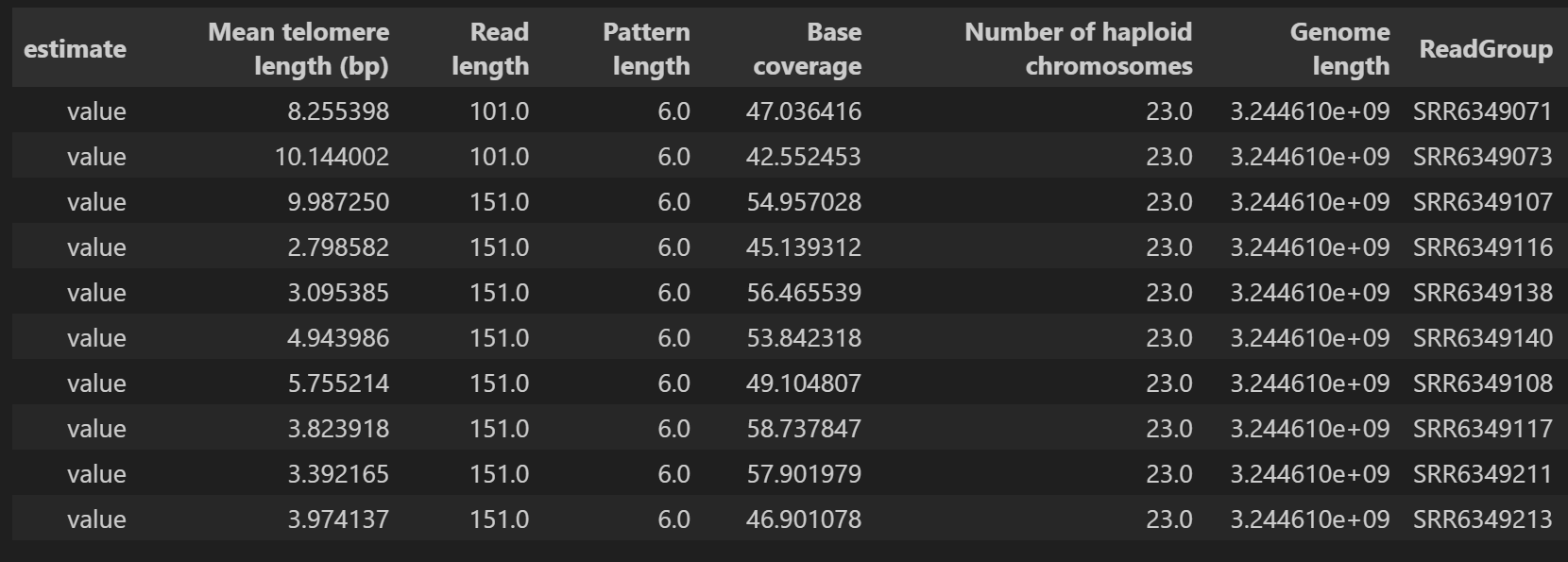
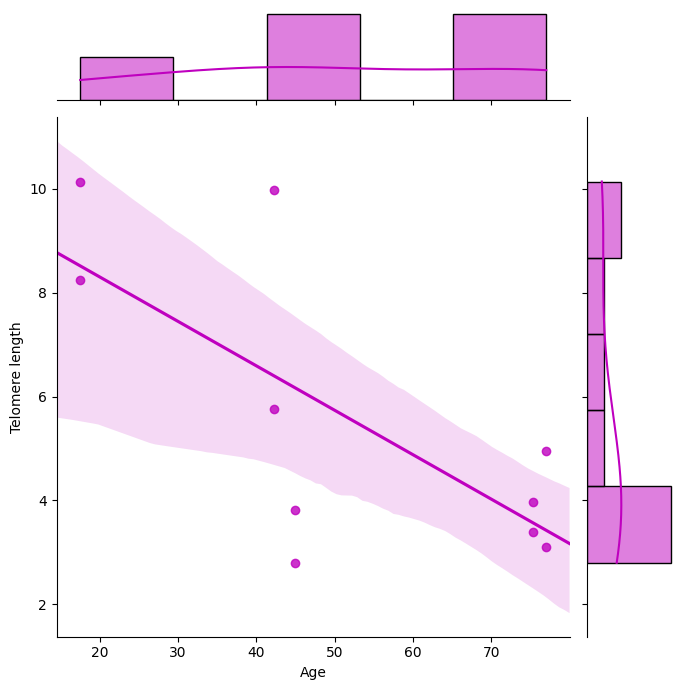
基于这个输出结果,我们可以利用merged_summary.csv文件中的Mean telomere length的数值来绘制估计的端粒长度与年龄之间的非线性相关关系。如下图所示.
以上便是本教程的全部内容了,希望你能在此教程中有所收获,能够学会如何正确下载,安装,编译和使用computel软件,并且估计输入fastq文件对应的端粒长度。