telseq的计算原理¶
总结来说,作者假定包含k个(k默认为7,可以在超参数中调整)TTAGGG(及其反向配对序列)重复的Reads就是来自端粒的Reads。
作者通过以下公式计算端粒长度:
$l = t_kSC$
公式中,$l$为端粒长度估算值,$t_k$是在阈值k下的端粒Reads的丰度。参数$S$由GC含量在48%到52%的Reads的数量,$C$是具有相同GC组成的基因组区域的累积长度。
telseq的安装与编译¶
telseq是一款根据全基因组测序数据(Genome Sequencing Data, BAMs)估算端粒长度的软件。
telseq由C++语言编写,在本教程中我们使用Linux环境运行telseq。
根据我的实际操作经验,运行telseq并不要求我们创建特定的运行环境。仅需要将工作路径切换到合适的位置并导入telseq所需的环境变量即可。
然而,在本教程中,我们将演示如何从零开始创建、安装、编译和使用telseq,以便提供一个全面的示例。
首先,我们会创建一个全新的环境,并将其命名为"telseq"。
# 创建新环境
conda create -n telseq python=3.8
# 启动环境
conda activate telseq
# 我们切换到一个空文件夹中,这个文件夹将存储Bamtools和telseq的代码文件。
cd /home/douyanmeiLab/hulei/WGS_test
# 下载和安装BamTools
git clone https://github.com/pezmaster31/bamtools.git
# 这里我们将BamTools下载到以下路径:/home/douyanmeiLab/hulei/WGS_test/bamtools
BamTools依赖于CMake进行文件构建,要求CMake的版本需要大于等于3.0。所以我们首先需要在命令行中输入以下代码,判断CMake版本。
如果CMake版本过低或者环境中缺少CMake,我们需要手动下载CMake。对于西湖大学高性能计算中心的用户来讲,使用命令行创建新环境后,默认会自带3.11.4版本的CMake(2023.10.8确认),所以不需要下载,直接使用即可。
# 确定CMake版本
cmake --version
当我们准备好了CMake之后,我们就可以来构建BamTools文件了。
首先我们切换到BamTools的顶级目录,并且在命令行中输入以下命令,以配置BamTools文件的构建过程。
cd /home/douyanmeiLab/hulei/WGS_test/bamtools
# DCMAKE_INSTALL_PREFIX是最终安装目录的根目录
cmake -DCMAKE_INSTALL_PREFIX=/home/douyanmeiLab/hulei/WGS/telomere/telseq/bamtools
接下来,我们在原路径上,在命令行中输入make以执行BamTools的实际构建过程。它基于前面由CMake生成的构建文件(通常是Makefile)来编译和链接项目的源代码。
make
最后,我们在命令行中输入以下命令,将已编译的程序或库文件安装到系统中。
make DESTDIR=/ install
我们可以在命令行中输入bamtools以判断BamTools的安装是否成功。
如果输出为下图的结果,则可以视为BamTools安装成功。
bamtools
# 我们切换回最高目录
cd /home/douyanmeiLab/hulei/WGS_test
# 下载telseq软件包。
git clone https://github.com/zd1/telseq.git
# 获取安装telseq所需的依赖项
conda install gxx_linux-64
conda install -c bioconda bamtools
接下来我们编译telseq文件。
具体来说,我们进入src目录并从该目录中运行autogen.sh以生成configure文件./autogen.sh,然后执行以下操作以生成可运行的二进制文件:
# 进入src目录
cd telseq
cd src
# 运行autogen.sh以生成configure文件
sh autogen.sh
# 生成可运行的二进制文件,由于bamtools没有安装在系统位置,我们需要手动指定bamtools的位置。
./configure --with-bamtools=/home/douyanmeiLab/hulei/WGS_test/bamtools
make
最后,我们需要切换到包含生成的二进制文件的路径,导入环境变量,然后直接输入telseq指令以判断能否使用telseq命令。
# 切换到可执行代码文件的目录。
cd /home/douyanmeiLab/hulei/WGS_test/telseq/src/Telseq
# 导入环境变量
export PATH=/home/douyanmeiLab/hulei/WGS_test/telseq/src/Telseq:$PATH
# 运行telseq指令
telseq
当运行telseq的结果为下图时,则恭喜你已经成功安装telseq了。
运行telseq以估计样品的平均端粒长度¶
你可以使用命令行或西湖大学高性能计算平台的srun系统来运行telseq。
下面提供了一个在srun系统中运行telseq的模拟代码示例。在这个示例中,我们将计算特定路径下所有的BAM文件的平均端粒长度,并将结果输出到output.csv文件中。
如果你想查找telseq工具包的其他参数及其默认参数,你可以在命令行中输入telseq,即可得到相关参数说明。
#!/bin/bash
#SBATCH -p intel-sc3,amd-ep2
#SBATCH -q normal
#SBATCH -J preprocess
#SBATCH -c 15
#SBATCH -o /home/douyanmeiLab/hulei/WGS/telomere/Final_output/log/telseq.log
#SBATCH --mem=60G
#SBATCH -t 120:00:00
cd /home/douyanmeiLab/hulei/WGS_test/telseq/src/Telseq
export PATH=/home/douyanmeiLab/hulei/WGS_test/telseq/src/Telseq:$PATH
telseq -o /home/douyanmeiLab/hulei/WGS/telomere/Final_output/telseq/output.csv \
/storage/douyanmeiLab/hulei/Data/Bam/*.bam
telseq结果说明¶
telseq的输出结果是一个CSV文件。对于这种类型的文件,你可以在Linux中手动打开它,或者创建一个IPython Notebook文件,并使用Pandas库来打开CSV文件。output.csv文件的内容如下:
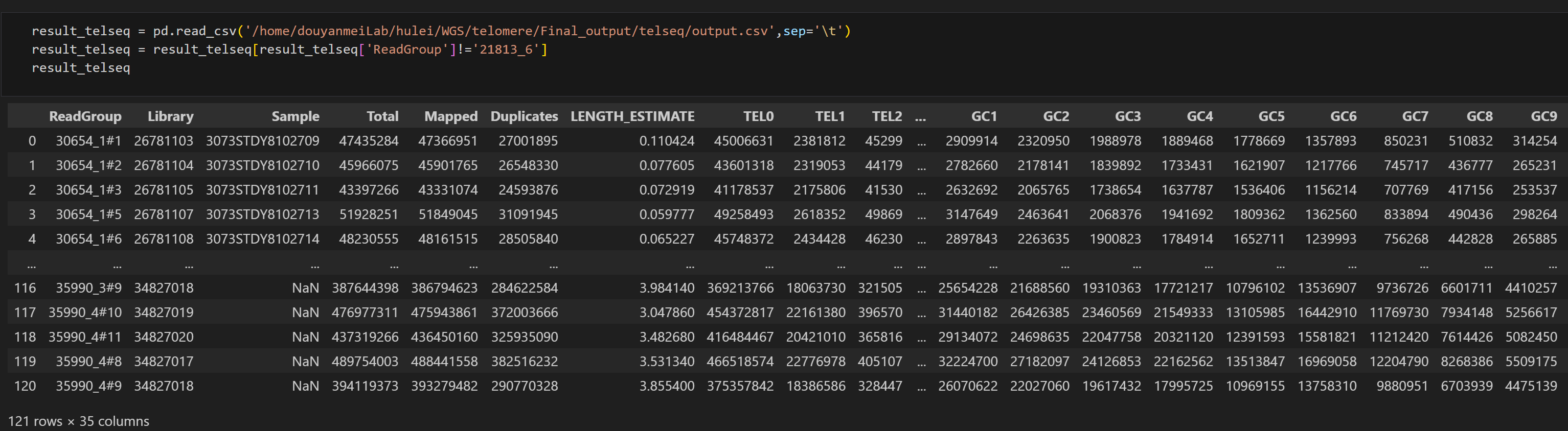
在此,我们简单介绍一下telseq输出文件中比较重要的几个结果。
ReadGroup:该参数由BAM头文件中的RG tag信息所确定,一般为样本的ID信息。
Total:在这个ReadGroup中所包含的全部Reads的总数。
Mapped:在这个ReadGroup中被成功映射到基因组的Reads的总数。
LENGH_ESTIMATE:telseq估计的端粒长度。
TEL0-TEL16:包含n(n的范围在0到16之间,且为整数)TTAGGG/CCCTAA重复序列的Reads总数。
GC1-GC9:GC比例在(40% + n * 2%)-(42% + (n + 1) * 2%)之间的Reads总数。
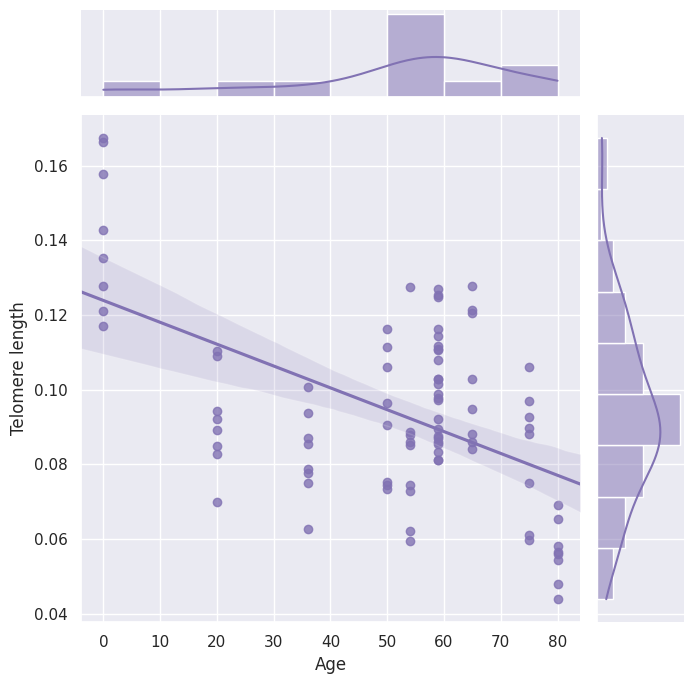
基于这个输出结果,我们可以利用output.csv文件中的LENGTH_ESTIMATE的数值来绘制估计的端粒长度与年龄之间的非线性相关关系。如下图所示.
以上便是本教程的全部内容了,希望你能在此教程中有所收获,能够学会如何正确下载,安装,编译和使用telseq软件,并且估计输入Bam文件对应的端粒长度。